
1. PCA优缺点
- 利用PCA达到降维目的,避免高维灾难。
- PCA把所有样本当作一个整体处理,忽略了类别属性,所以其丢掉的某些属性可能正好包含了重要的分类信息
2. PCA原理
条件1:给定一个m*n的数据矩阵D, 其协方差矩阵为S. 如果D经过预处理, 使得每个每个属性的均值均为0, 则有S=DTDS=DTD。
PCA的目标是找到一个满足如下性质的数据变换:
- 每对不同的新属性的协方差为0,即属性间相互独立;
- 属性按照每个属性捕获的数据方差大小进行排序;
- 第一个属性捕获尽可能多的数据方差;
- 满足正交性的前提下,每个属性尽可能多的捕获剩余方差。
条件2:由于协方差矩阵是半正定矩阵,则其具有非负特征值。令λ1,λ2,...,λnλ1,λ2,...,λn是S的特征值,并且可以排序,保证λ1≥λ2≥...≥λnλ1≥λ2≥...≥λn.令U为S的特征向量矩阵。
因此,有:
- 数据矩阵D′=DUD′=DU是变换后的数据集,满足上述条件;
- 第ii个属性的方差是λiλi;
- 原属性的方差和等于新属性的方差和;
- 新属性排序即为主成分排序;
通常情况下,我们排序较后的主成分其实为次要成分,基于降维目的,只需选取前m个主成分作为新属性即可。
3. R中使用PCA
程序
PCA的目标是用一组较少的不相关变量代替大量相关变量,同时尽可能保留初始变量的信息,这些推导所得的变量称为主成分,它们是观测变量的线性组合。如第一主成分为:
PC1=a1X1=a2X2+……+akXk 它是k个观测变量的加权组合,对初始变量集的方差解释性最大。
#1.载入并清洗处理数据
read.table()
ovun.sample()
#2.作主成分分析并显示分析结果
#princomp()主成分分析 可以从相关阵或者从协方差阵做主成分分析
#cor是逻辑变量当cor=TRUE表示用样本的相关矩阵R做主成分分析
#当cor=FALSE表示用样本的协方差阵S做主成分分析
test.pr<-princomp(dbboth,cor=TRUE)
#summary()提取主成分信息
#loading是逻辑变量当loading=TRUE时表示显示loading 的内容
#loadings的输出结果为载荷是主成分对应于原始变量的系数即Q矩阵
summary(test.pr,loadings=TRUE)
# 分析结果含义
# #----Standard deviation 标准差 其平方为方差=特征值
# #----Proportion of Variance 方差贡献率
# #----Cumulative Proportion 方差累计贡献率
#4.画主成分的碎石图并预测
screeplot(test.pr,type="lines")
p<-predict(test.pr)
p

#biplot()画出数据关于主成分的散点图和原坐标在主成分下的方向
主成分分析(PCA)是一种数据降维技巧,它能将大量相关变量转化为一组很少的不相关变量,这些无关变量称为主成分。
探索性因子分析(EFA)是一系列用来发现一组变量的潜在结构的方法,通过寻找一组更小 的、潜在的或隐藏的结构来解释已观测到的、变量间的关系。
1.R中的主成分和因子分析
R的基础安装包中提供了PCA和EFA的函数,分别为princomp ()和factanal()
psych包中有用的因子分析函数
函数 | 描述 |
principal() | 含多种可选的方差放置方法的主成分分析 |
fa() | 可用主轴、最小残差、加权最小平方或最大似然法估计的因子分析 |
fa.parallel() | 含平等分析的碎石图 |
factor.plot() | 绘制因子分析或主成分分析的结果 |
fa.diagram() | 绘制因子分析或主成分分析的载荷矩阵 |
scree() | 因子分析和主成分分析的碎石图 |
PCA/EFA 分析流程:
(1)数据预处理;PCA和EFA都是根据观测变量间的相关性来推导结果。用户可以输入原始数据矩阵或相关系数矩阵列到principal()和fa()函数中,若输出初始结果,相关系数矩阵将会被自动计算,在计算前请确保数据中没有缺失值;
(2)选择因子分析模型。判断是PCA(数据降维)还是EFA(发现潜在结构)更符合你的分析目标。若选择EFA方法时,还需要选择一种估计因子模型的方法(如最大似然估计)。
(3)判断要选择的主成分/因子数目;
(4)选择主成分/因子;
(5)旋转主成分/因子;
(6)解释结果;
(7)计算主成分或因子得分。
2.主成分分析
PCA的目标是用一组较少的不相关变量代替大量相关变量,同时尽可能保留初始变量的信息,这些推导所得的变量称为主成分,它们是观测变量的线性组合。如第一主成分为:
PC1=a1X1=a2X2+……+akXk 它是k个观测变量的加权组合,对初始变量集的方差解释性最大。
第二主成分是初始变量的线性组合,对方差的解释性排第二, 同时与第一主成分正交(不相关)。后面每一个主成分都最大化它对方差的解释程度,同时与之前所有的主成分都正交,但从实用的角度来看,都希望能用较少的主成分来近似全变量集。
(1)判断主成分的个数
PCA中需要多少个主成分的准则:
根据先验经验和理论知识判断主成分数;
根据要解释变量方差的积累值的阈值来判断需要的主成分数;
通过检查变量间k*k的相关系数矩阵来判断保留的主成分数。
最常见的是基于特征值的方法,每个主成分都与相关系数矩阵的特征值 关联,第一主成分与最大的特征值相关联,第二主成分与第二大的特征值相关联,依此类推。
Kaiser-Harris准则建议保留特征值大于1的主成分,特征值小于1的成分所解释的方差比包含在单个变量中的方差更少。
Cattell碎石检验则绘制了特征值与主成分数的图形,这类图形可以展示图形弯曲状况,在图形变化最大处之上的主成分都保留。
最后,还可以进行模拟,依据与初始矩阵相同大小的随机数矩阵来判断要提取的特征值。若基于真实数据的某个特征值大于一组随机数据矩阵相应的平均特征值,那么该主成分可以保留。该方法称作平行分析。
利用fa.parallel()函数,可同时对三种特征值判别准则进行评价。
-
library(psych) -
fa.parallel(USJudgeRatings[,-1],fa="PC",n.iter=100,show.legend=FALSE,main="Screen plot with parallel analysis")
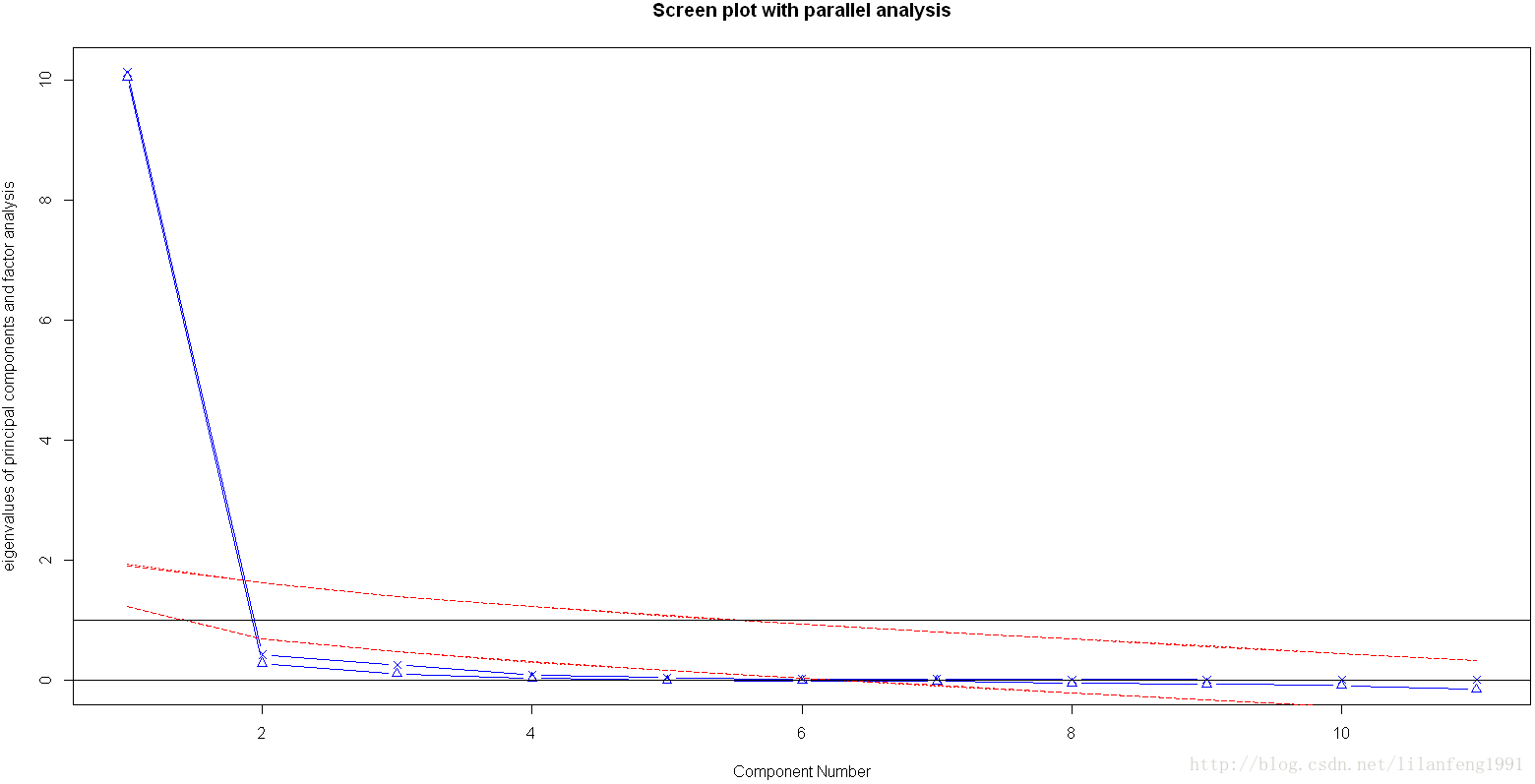
碎石头、特征值大于1准则和100次模拟的平行分析(虚线)都表明保留一个主成分即可保留数据集的大部分信息,下一步是使用principal()函数挑选出相应的主成分。
(2)提取主成分
principal()函数可根据原始数据矩阵或相关系数矩阵做主成分分析
格式为:principal(的,nfactors=,rotate=,scores=)
其中:r是相关系数矩阵或原始数据矩阵;
nfactors设定主成分数(默认为1);
rotate指定旋转的方式[默认最大方差旋转(varimax)]
scores设定是否需要计算主成分得分(默认不需要)。
-
美国法官评分的主成分分析 -
library(psych) -
pc<-principal(USJudgeRatings[,-1],nfactors=1) -
pc
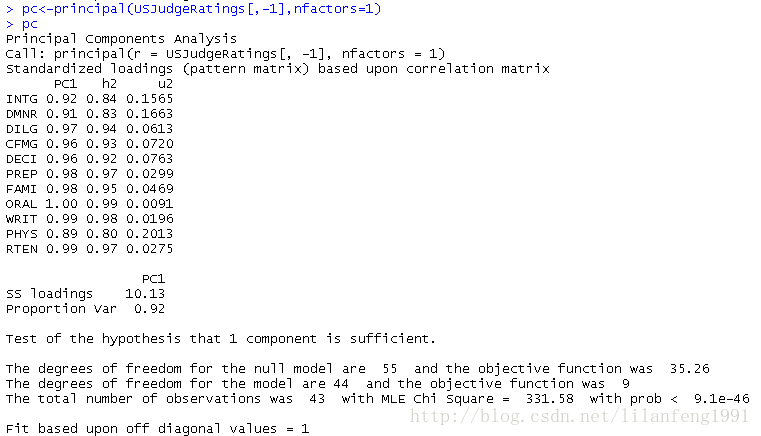
此处,输入的是没有ONT变量的原始,并指定获取一个未旋转的主成分。由于PCA只对相关系数矩阵进行分析,在获取主成分前,原始数据将会被自动转换为相关系数矩阵。
PC1栏包含了成分载荷,指观测变量与主成分的相关系数。如果提取不止一个主成分,则还将会有PC2、PC3等栏。成分载荷(component loadings)可用来解释主成分的含义。此处可看到,第一主成分(PC1)与每个变量都高度相关,也就是说,它是一个可用来进行一般性评价的维度。
h2柆指成分公因子方差-----主成分对每个变量的方差解释度。
u2栏指成分唯一性-------方差无法 被主成分解释的比例(1-h2)。
SS loadings行包含了主成分相关联的特征值,指的是与特定主成分相关联的标准化后的方差值。
Proportin Var行表示的是每个主成分对整个数据集的解释程度。
结果不止一个主成分的情况
-
library(psych) -
fa.parallel(Harman23.cor$cov,n.obs=302,fa="pc",n.iter=100,show.legend=FALSE,main="Scree plot with parallel analysis")
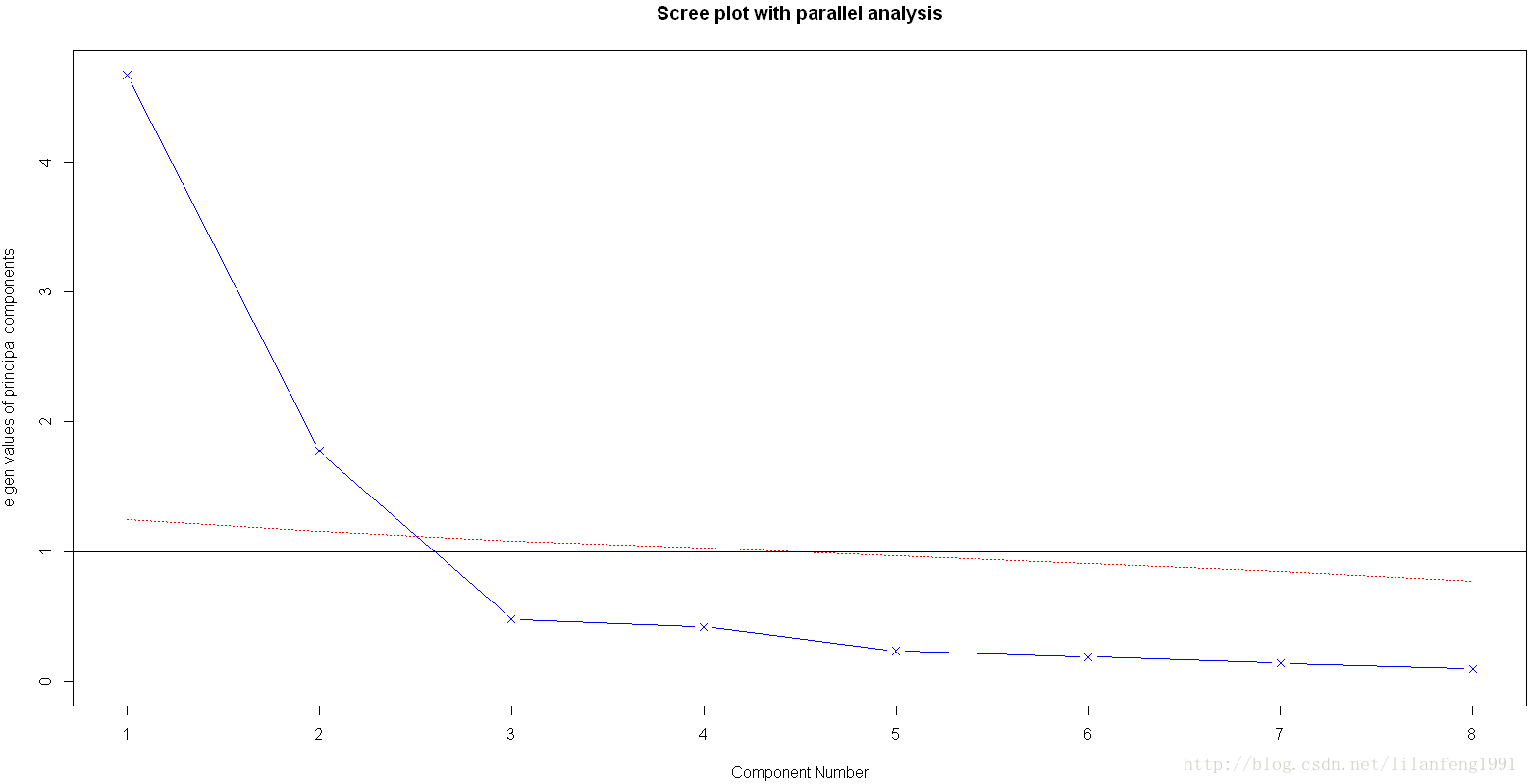
载荷阵解释了成分和因子的含义,第一成分与每个身体测量指标都正相关,看起来似乎是一个一般性的衡量因子;第二主成分与前四个变量负相关,与后四个变量正相关,因此它看起来似乎是一个长度容量因子。但理念上的东西都不容易构建,当提取了多个成分时,对它们进行旋转可使结果更具有解释性。
(3)主成分旋转
旋转是一系列将成分载荷阵变得更容易解释的数学方法,它们尽可能地对成分去噪。
旋转方法有两种:使选择的成分保持不相关(正效旋转),和让它们变得相关(斜交旋转)。
旋转方法也会依据去噪定义的不同而不同。
最流行的下次旋转是方差极大旋转,它试图对载荷阵的列进行去噪,使得每个成分只是由一组有限的变量来解释(即载荷阵每列只有少数几个很大的载荷,其他都是很小的载荷)。
-
install.packages("GPArotation") -
library(GPArotation) -
rc<-principal(Harman23.cor$cov,nfactors=2,rotate="varimax") -
rc
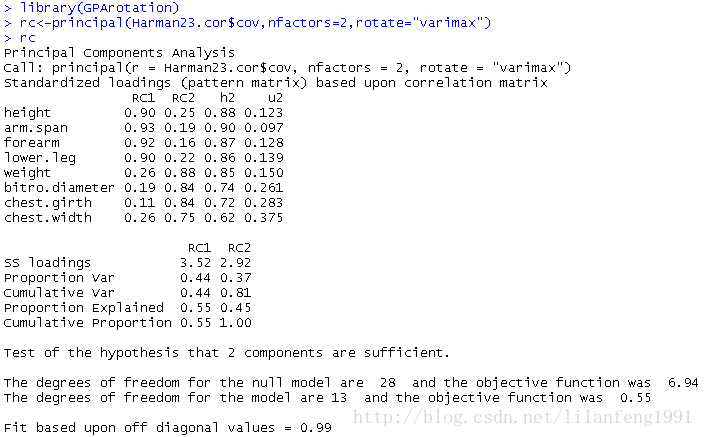
列名从PC变成了RC,以表示成分被旋转
观察可以发现第一主成分主要由前四个变量来解释,第二主成分主要由变量5到变量8来解释。
注意两个主成分仍不相关,对变量的解释性不变,这是因为变量的群组没有发生变化。另外,两个主成分放置后的累积方差解释性没有变化,变的只是各个主成分对方差的解释(成分1从58%变为44%,成分2从22%变为37%)。各成分的方差解释度趋同,准确来说,此时应该称它们为成分而不是主成分。
(4)获取主成分得分
利用principal()函数,很容易获得每个调查对象在该主成分上的得分。
<strong>从原始数据中获取成分得分</strong>-
library(psych) -
pc<-principal(USJudgeRatings[,-1],nfactors=1,score=TRUE) -
head(pc$scores)
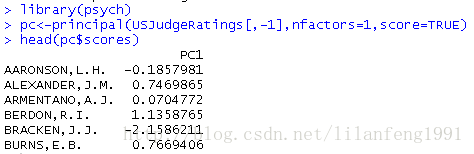
当scores=TRUE时,主成分得分存储在principal()函数返回对象的scores元素中。
cor(USJudgeRatings$CONT,PC$scores)
<strong>获取主成分得分的系数</strong>-
library(psych) -
rc<-principal(Harman23.cor$cov,nfactor=2,rotate="varimax") -
round(unclass(rc$weights),2)

得到主成分得分:
PC1=0.28*height+0.30*arm.span+0.30*forearm+0.29*lower.leg-0.06*weight-0.08*bitro.diameter-0.10*chest.girth-0.04*chest.width
PC2=-0.05*height-0.08*arm.span-0.09*forearm-0.06*lower.leg+0.33*weight+0.32*bitro.diameter+0.34*chest.girth+0.27*chest.width
3.探索性因子分析
EFA的目标是通过发掘隐藏在数据下的一组较少的、更为基本的无法观测的变量,来解释一组可观测变量的相关性。这些虚拟的、无法观测的变量称作因子。(每个因子被认为可解释多个观测变量间共有的方差,也叫作公共因子)
模型的形式为:
Xi=a1F1+a2F2+……apFp+Ui
Xi是第i个可观测变量(i=1,2,……k)
Fj是公共因子(j=1,2,……p)
并且p<k
-
options(digits=2) -
covariances<-ability.cov$cov -
correlations<-cov2cor(covariances) -
correlations
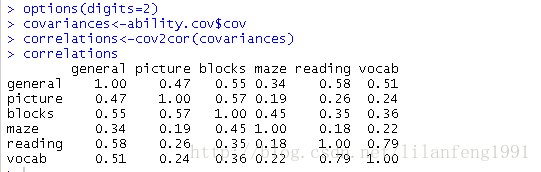
ability.cov提供了变量的协方差矩阵
cov2cor()函数将其转化为相关系数矩阵
(1)判断需提取的公共因子数
-
library(psych) -
convariances<-ability.cov$cov -
correlations<-cov2cor(covariances) -
fa.parallel(correlations,n.obs=112,fa="both",n.iter=100,main="Scree plots with parallel analysis")
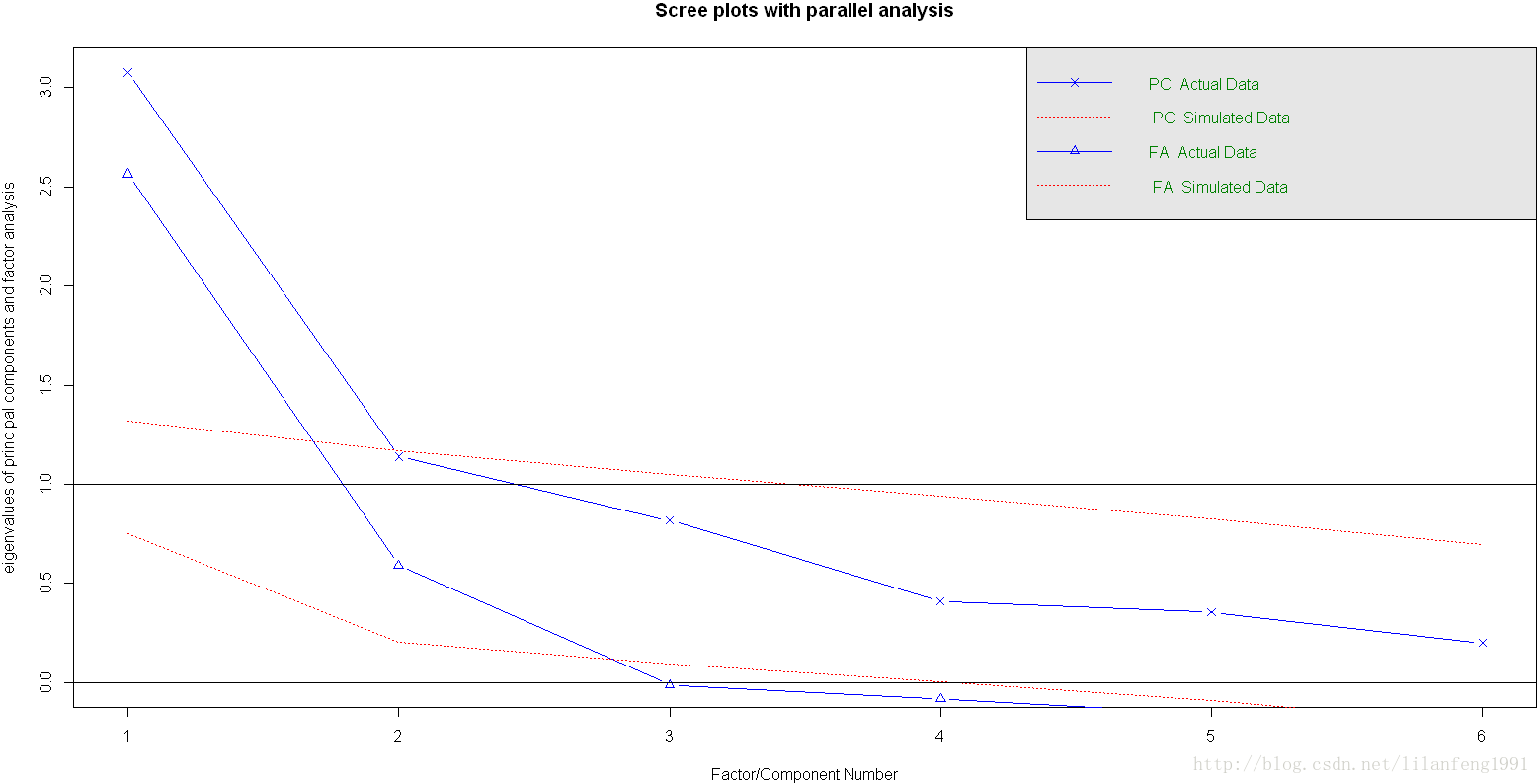
若使用PCA方法,可能会选择一个成分或两个成分。当摇摆不定时,高估因子数通常比低估因子数的结果好,因为高估因子数一般较少曲解“真实”情况。
(2)提取公共因子
可使用fa()函数来提取因子
fa()函数的格式为:
fa(r,nfactors=,n.obs=,rotate=,scores=,fm)
r是相关系数矩阵或原始数据矩阵;
nfactors设定提取的因子数(默认为1);
n.obs是观测数(输入相关系数矩阵时需要填写);
rotate设定放置的方法(默认互变异数最小法);
scores设定是否计算因子得分(默认不计算);
fm设定因子化方法(默认极小残差法)。
与PCA不同,提取公共因子的方法很多,包括最大似然法(ml)、主轴迭代法(pa)、加权最小二乘法(wls)、广义加权最小二乘法(gls)和最小残差法(minres)。
<strong>未旋转的主轴迭代因子法</strong>-
fa<-fa(correlations,nfactors=2,rotate="none",fm="pa") -
fa
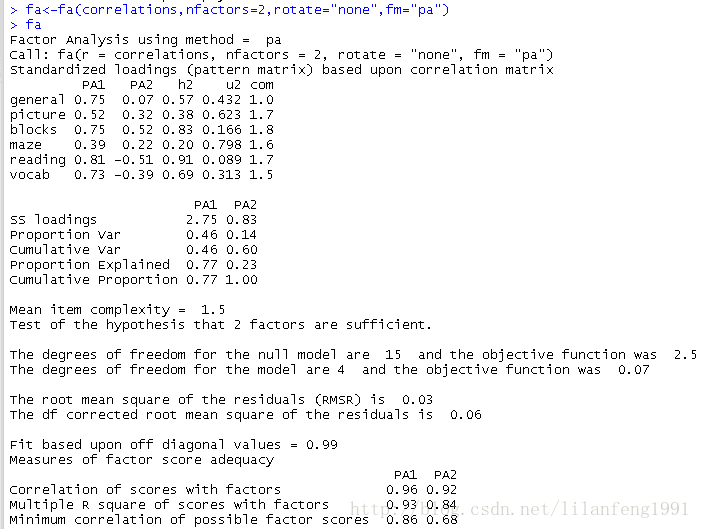
(3)因子旋转
<strong>用正交旋转提取因子</strong>-
fa.varimax<-fa(correlations,nfactors=2,rotate="varimax",fm="pa") -
fa.varimax
<strong>正交放置将人为地强制两个因子不相关</strong><strong>正交旋转,因子分析的重点在于因子结构矩阵(变量与因子的相关系数)</strong>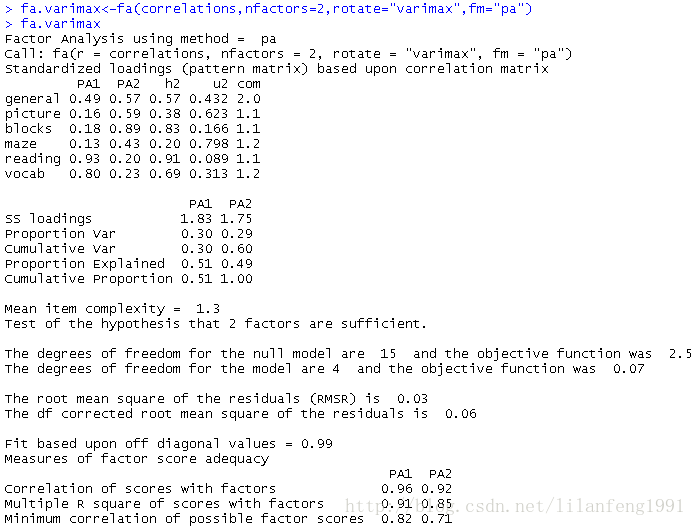
用斜交旋转提取因子
-
fa.promax<-fa(correlations,nfactors=2,rotate="promax",fm="pa") -
fa.promax
<strong>对于斜交旋转,因子分析会考虑三个矩阵:因子结构矩阵、因子模式矩阵和因子关联矩阵</strong><strong>因子模式矩阵即标准化的回归系数矩阵,它列出了因子的预测变量的权重;</strong><strong>因子关联矩阵即因子相关系数矩阵;</strong><strong>因子结构矩阵(或称因子载荷阵),可使用公式F=P*Phi来计算得到,其中F是载荷阵,P为因子模式矩阵,Phi为因子关联矩阵。</strong>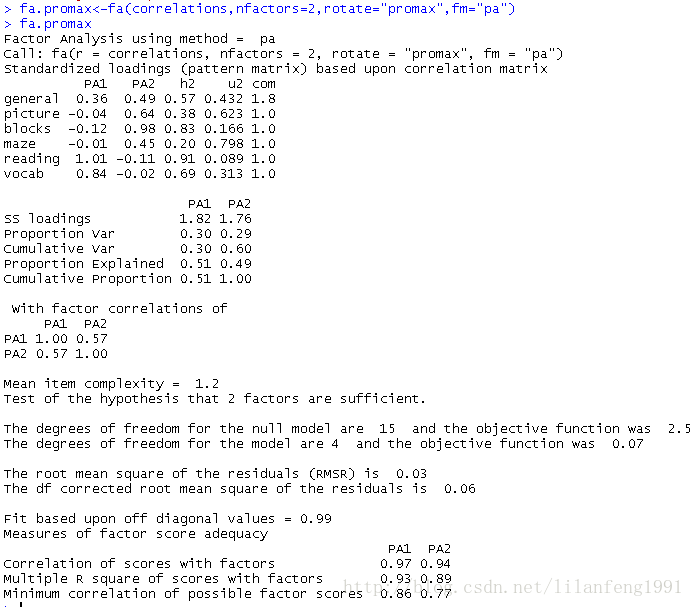
1. fsm<-function(oblique){
2.
if(class(oblique)[2]=="fa"&is.null(oblique$Phi)){
3.
warning("Object doesn't look like oblique EFA")
4.
}else{
5.
P<-unclass(oblique$loading)
6.
F<-P%*%oblique$Phi
7.
colnames(F)<-c("PA1","PA2")
8.
return (F)
9.
}
10.
}
11.
fsm(fa.promax)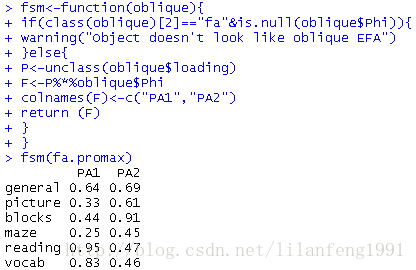
可以看到变量与因子间的相关系数。将它们与正交旋转所得因子载荷阵相比,发现该载荷阵列的噪音较大,这是因为之前允许潜在因子相关。虽然斜交方法更为复杂,但模型将更加符合真实数据。
使用factor.plot()或fa.diagram()函数,可绘制正交或斜交结果的图形
factor.plot(fa.promax,labels=rownames(fa.promax$loadings))
fa.diagram(fa.promax,simple=TRUE)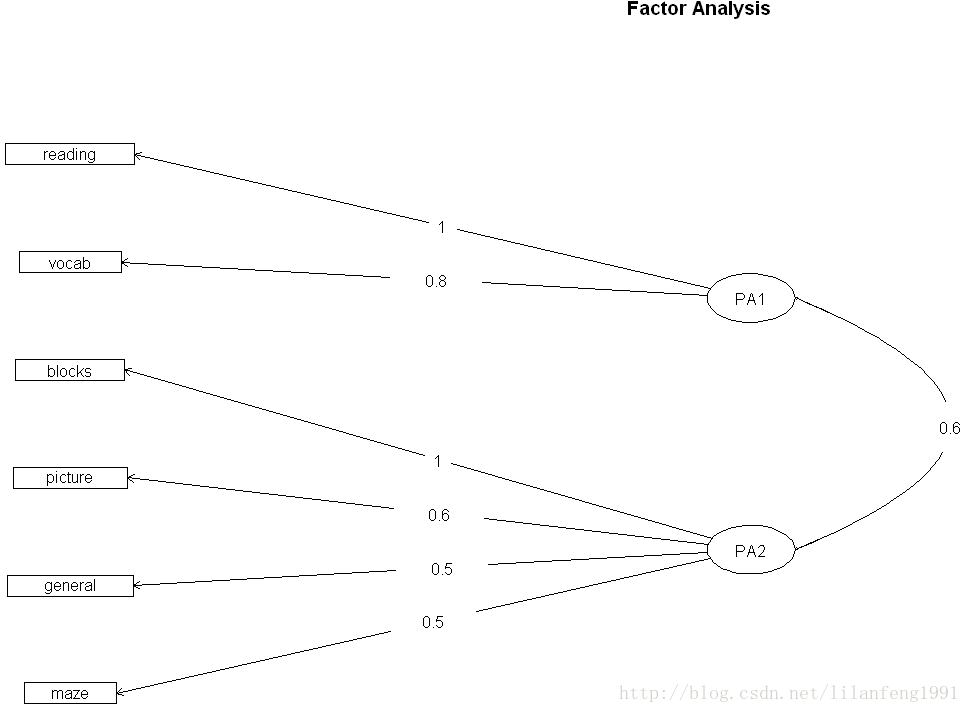
(4)因子得分
EFA并不十分关注因子得分,在fa()函数中添加score=TRUE选项,便可轻松地得到因子得分。另外还可以得到得分系数(标准化的回归权重),它在返回对象的weights元素中。
fa.promax$weights
4.其他
(1)
对因子分析非常有用的软件包,FactoMineR包不仅提供了PCA和EFA方法,还包含潜变量模型。
FAiR包使用遗传算法来估计因子分析模型,增强了模型参数估计能力,能够处理不等式的约束条件;
GPArotation包提供了许多因子旋转方法
nFactors包,提供了用来判断因子数目方法。
(2)其他潜变量模型
先验知识的模型:先从一些先验知识开始,比如变量背后有几个因子、变量在因子上的载荷是怎样的、因子间的相关性如何,然后通过收集数据检验这些先验知识。这种方法称作验证性因子分析(CFA)。 做CFA的软件包:sem、openMx和lavaan等。
ltm包可以用来拟合测验和问卷中各项目的潜变量模型。
潜类别模型(潜在的因子被认为是类别型而非连续型)可通过FlexMix、lcmm、randomLCA和poLC包进行拟合。lcda包可做潜类别判别分析,而lsa可做潜在语义分析----一种自然语言处理中的方法。ca包提供了可做简单和多重对应分析的函数。
R中还包含了众多的多维标度法(MDS)计算工具。MDS即可用发现解释相似性和可测对象间距离的潜在维度。
cmdscale()函数可做经典的MDS
MASS包中的isoMDS()函数可做非线性MDS
vagan包中则包含了两种MDS的函数


















