
介绍
在分子生物学中,DNA 和蛋白质可以表示为字母序列。
DNA 序列由 A、T、G、C 组成,代表核苷碱基(nucleobases) 腺嘌呤、胸腺嘧啶、鸟嘌呤和胞嘧啶。
蛋白质由 20 个不同的字母组成,表示 20 种不同的氨基酸。
比较来自同一生物体或来自不同生物体的两个序列,称为 序列比较 (Sequence comparison),是分子生物学中的一项重要任务。
它有助于为许多生物学问题提供解决方案,例如:
- 预测蛋白质的结构和功能
- 推断物种的进化历史和相关性
- 定位基因/蛋白质中的常见子序列以识别常见基序,
- 作为 DNA 测序的基因组组装中的一个子问题
为了进行序列比较,我们首先进行 序列比对(sequence alignment)。
什么是序列比对?
序列比对是将一个序列置于另一个序列之上以识别相似字符或子串之间的对应关系的一种方式。
它可以在脱氧核糖核酸 (DNA)、核糖核酸 (RNA) 或蛋白质序列上进行。
来自不同生物体的序列可能具有不同的大小。对齐需要在序列的任意位置插入空格,以便两者具有相同的大小。
在序列的开头或结尾插入“空格”或“间隙”。
让我们考虑一个例子。 假设我们有以下两个 DNA 序列 GACGGATTAG 和 GATCGGAATAG。
这两个序列之间的一种可能比对如下所示
GA-CGGATTAG
|| |||| |||
GATCGGAATAG请注意根据此比对的两个序列的差异。 第二个序列中的额外 T,以及从 T 到 A 的替换。在第一个序列的合适位置添加一个“空格”以对齐序列。
比对类型
根据我们正在寻找的内容,有两种类型的比对方式:
全局比对( Global alignmen ):
沿序列的整个长度将查询序列与目标序列对齐。 全局比对是跨越两个查询序列全长的全局优化过程。
局部比对( Local alignment ):
将查询序列的子串与目标序列的子串对齐,即找到两个序列之间相似度高的局部区域。
根据被比对的序列数量,也有两种比对方式:
双序列比对( pairwise alignment ):
涉及两个序列,查询和与之对齐的目标。
多序列比对( multiple alignment ):
涉及两个以上序列的比对。
序列比对的软件工具
定位查询序列与数据库序列的相似区域是生物信息学中的一项具有挑战性的任务。
BLAST 和 FASTA 等工具有助于检测生物体之间的相似区域。它涉及使用局部成对比对、详尽的启发式算法和动态规划方法(如 Smith-Waterman 算法)来检测查询序列与数据库序列的相似区域。
最长公共子序列 (LCS) 问题
检测两个或多个序列相似性的一种方法是找到它们的最长公共子序列(Longest common subsequence)。
两个序列的最长公共子序列 (LCS) 是两个序列共有的具有最大可能长度的子序列。
举例
让我们考虑一个例子。 假设我们有以下两个 DNA 序列:TAGTCACG 和 AGACTGTC。
两个序列的LCS是AGACG,可以从下面的比对中得到。
TAGTCAC-G--
|| || |
-AG--ACTGTC还有其他可能的较短长度的公共/共有序列 ( common sequences ),例如 AGCG 或 AGTC。
尽管通常多个 LCS 是可能的,但对于这个特定示例只有一个 LCS,即这两个序列没有其他长度为 5 的公共子序列。
LCS 还有助于计算两个序列的相似程度:LCS 越长,相似性越高。
LCS算法
假设我们有两个长度分别为 m 和 n 的序列 S1 和 S2,其中
S1 = a1 a2 … am
S2 = b1 b2 … bn。
我们将构造一个矩阵 A,其中 Ai,j 表示 a1 a2 … ai 和 b1 b2 … bj 的最长公共子序列的长度。

序列 S1 =
TAGTCACG和 S2 =AGACTGTC的示例矩阵。
序列 S1 垂直写入,S2 水平写入。 将代表行的每个字母与代表列的每个字母进行比较。 我们将逐行、逐列进行。
- 如果 ai = bj,那么我们找到了匹配项。 当前匹配的得分为 1,而 Ai-1,Bj-1 来自我们已经从子串 a1 … ai-1 和 b1 … bj-1 获得的 LCS 的其余部分。
- 如果 ai ≠ bj,那么我们有一个不匹配。 在这种情况下,我们需要考虑两种可能性。 LCS a1 … ai 和 b1 … bj-1 ,以及 a1 … ai-1 和 b1 … bj 的 LCS。
因此,我们有
并且 A0,0 = A0,j = Ai,0 = 0 对于 1 ≤ i ≤ m,1≤ j ≤ n。
填充 m × n 矩阵 A 需要 O(nm) 次。这种方法称为 动态规划( dynamic programming )
Step by step 例子
矩阵逐行、逐列填充。
- 第一行 R1 和列 C1 填充为零。
- (不匹配示例)将 R2 行中的第一个字母与 C2 列进行比较。在这种情况下,单元格包含“T”和“A”。由于是不匹配,所以从左边或上面取分数,即(R1,C2)和(R2,C1)。由于两个值都是“0”,因此在这种情况下,总分变为 max(0, 0) = 0。
- 我们继续填充 R2 的其余部分。
- (匹配示例)当 R3 与 C2 进行比较时,两个单元格都包含“A”。由于这是一场比赛,因此得分为 1 + 对角线上方和左侧的得分,即 (R2, C1),在这种情况下为“0”。所以总分变为1。
- 我们继续填充 R3 的其余部分。
- (匹配示例) 类似地,当 R4 与 C3 进行比较时,它再次匹配。这次的分数是 1 + (R3, C2) 的分数,即 1。因此,总分变为 2。
- 整个矩阵就是这样填充的。最终分数由 Am,n = 5 给出。
序列 S1 =
TAGTCACG和 S2 =AGACTGTC的示例矩阵。
回溯法(Traceback)
实际的子序列是通过执行回溯推导出来的,即从右下角到左上角向后跟踪。 当长度在所有方向上减少时,序列必须匹配。
当一个单元格中可能有两个箭头时,可能有多个路径(在下面的框中给出)。
在这种情况下,可以选择任一路径。

对角边的位置包含构建 LCS 所需的所有信息,以及知道 LCS 对应的 S1 和 S2 中的位置。
上例的 LCS 是长度为 5 的 AGACG。
S1 和 S2 的斜边位置下划线如下:TAGTCACG 和 AGACTGTC。
打分矩阵(Scoring Matrices)
在上面的例子中,得到一个不匹配的 0 和一个匹配的 1。
这可以推广到每个字母表的删除、插入或匹配的不同分数,以及每对可能的不匹配字母表的不同分数。

DNA 比对的评分矩阵示例。
矩阵包括匹配和不匹配的分数。
单位矩阵等效于上面讨论的 LCS 算法。
匹配和错配的得分基于碱基(在 DNA 的情况下)和氨基酸(在蛋白质的情况下)的进化重要性。
例如,根据经验观察到的氨基酸出现的保守性质,赋予氨基酸权重。
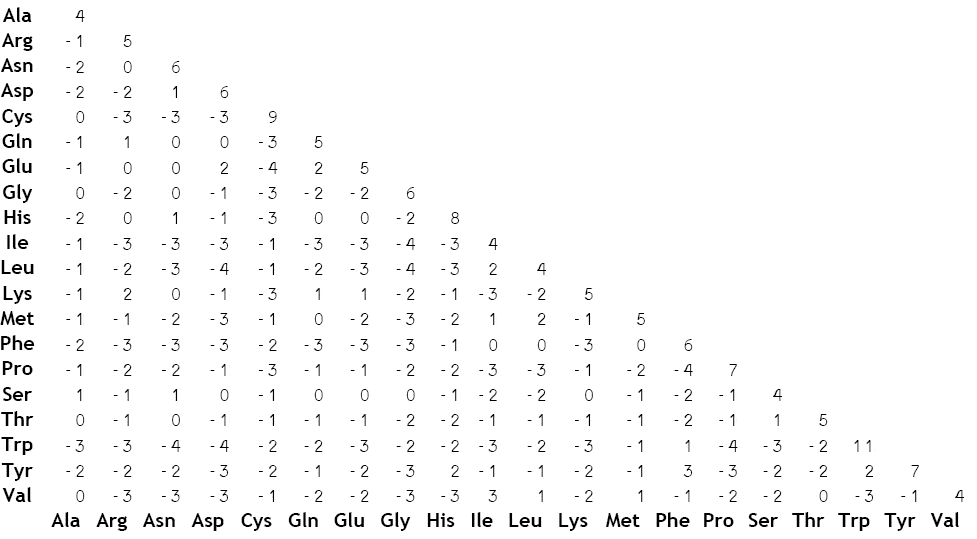
BLOSUM(BLOcks SUbstitution Matrix )矩阵
蛋白质序列比对常用的评分矩阵。每一行、每一列是一个氨基酸,一个蛋白质是一个氨基酸序列
≥0 的得分代表对应的一对氨基酸为相似氨基酸,<0 的是不相似的氨基酸
BLOSUM 后面跟一个小数字的矩阵适合用于比较相似度低的序列,也就是亲缘关系远的序列;
BLOSUM 后面跟一个大数字的矩阵适合比较相似度高的序列,也就是亲缘关系近的序列。


















